neuralgap.io
It’s quite clear by now that deep learning shows incredible promise in predicting protein-ligand interactions. However, their success is still fundamentally constrained by challenges spanning multiple domains: from dynamic nature of proteins and nucleic acids, binding pocket diversity, compound library biases, to selecting appropriate model architectures and evaluation frameworks. These challenges can be seen across different scales – from atomic-level flexibility to system-wide conformational changes – and across different aspects of the drug discovery pipelines, from target protein representation to ligand diversity considerations. In this series, we systematically explore these interconnected challenges, beginning with a detailed examination of protein-ligand representation problems that continue to test even our most sophisticated AI systems.
In this first article of our series, we examine how machine learning-based scoring functions face fundamental challenges in virtual screening due to the complex nature of protein representation. At the heart of these challenges lies the need for scoring functions to capture both the dynamic nature of proteins and the complexities of binding site interactions. Current AI architectures must grapple with representing structural flexibility across multiple scales – from atomic vibrations to domain movements – while simultaneously accounting for the diverse characteristics of binding pockets. Our analysis further explores how regulatory elements like post-translational modifications and allosteric sites compound these representation challenges.
Structural flexibility and dynamics of proteins present fundamental computational challenges that complicate their representation in AI systems. The core difficulty lies in capturing the multi-scale nature of protein motion – spanning from atomic fluctuations to large conformational shifts. These movements occur across various timescales, from picoseconds to milliseconds, and can involve thousands of atoms moving in coordinated patterns.
The representation challenge is further compounded by the interdependent nature of protein movements. Local changes in side-chain conformations can trigger larger domain movements, creating complex cause-and-effect relationships that span both spatial and temporal dimensions. Additionally, these structural changes often follow multiple possible pathways rather than a single trajectory, creating a branching tree of potential conformational states. This multiplicity of possible states exponentially increases the dimensionality of the problem, making it difficult to create comprehensive training datasets that capture all relevant protein configurations.
Perhaps the most significant challenge lies in protein flexibility. The same protein region may exhibit different dynamic behaviors depending on factors such as ligand binding, post-translational modifications, or other physiological conditions. This context-dependency means that static structural data, even when extensive, may fail to capture the true complexity of the system. Furthermore, the energy landscapes governing these conformational changes are often rugged and complex, with multiple local minima and transition states that are difficult to characterize. This creates a fundamental challenge in predicting not just the possible conformational states, but also the likelihood and pathways of transitions between them.
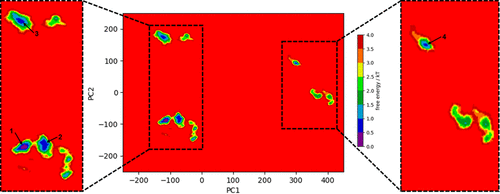
Image of free energy minima
To better understand the multi-scale challenges in representing protein dynamics, Table 1 breaks down key structural features into their static and dynamic components. This classification highlights how each protein element – from backbone atoms to entire domains – requires different mathematical frameworks for representation, while also noting specific computational hurdles that arise at each level.
Table 1: How structural features (static vs. dynamic) manifest in inputs
| Feature Type | Static Representation | Dynamic/Temporal Representation | Challenges in Representation |
| Backbone atoms | 3D coordinates (x,y,z) | Time series of coordinates (x,y,z,t) | Variable sequence lengths |
| Side chains | Rotamer states | Trajectory of torsion angles | Multiple possible conformations |
| Domain movements | Fixed domain positions | Time-dependent displacement vectors | Large-scale coordinated motions |
Beyond general protein dynamics, binding pocket diversity presents a fundamental challenge in computational drug discovery due to the extreme heterogeneity in their physical and chemical characteristics. The primary complexity arises from the vast spectrum of pocket geometries – ranging from deep, tunnel-like cavities to shallow surface grooves, and from small, defined pockets to large, amorphous binding regions. This geometric variability creates an immediate challenge in developing standardized representations that can meaningfully capture such diverse spatial arrangements while maintaining comparative analysis capabilities across different protein systems.
The dynamic nature of binding pockets adds another layer of complexity to their characterization. Pockets are not static entities but rather flexible regions that can expand, contract, or even transiently appear and disappear. This plasticity manifests in various ways – from minor side-chain movements to major backbone rearrangements that significantly alter pocket volume and shape. The challenge extends beyond merely accounting for different sizes; it involves understanding how these changes affect the pocket’s chemical environment, including hydrogen bond donors/acceptors shifts, hydrophobic surfaces, and electrostatic fields. Moreover, the presence of structural water molecules, which can be either displaced or incorporated into binding interactions, creates additional complexity in defining pocket boundaries and characteristics.
Beyond structural variations, the microenvironment within binding pockets presents distinct challenges for AI representation schemes. Each pocket possesses a unique combination of amino acid residues that create distinct physicochemical properties – including electrostatic gradients, hydrophobic patches, and specific geometric constraints – which must be encoded in ways that neural networks can effectively process. These properties form complex, non-uniform 3D patterns that vary significantly between protein families, challenging traditional AI featurization approaches that often assume spatial invariance. The context-dependent and non-additive nature of these properties particularly challenges deep learning models, as they must learn to recognize how different physicochemical features combine in non-linear ways to influence binding affinity. This complexity makes it difficult to develop universal scoring functions or feature representations that generalize across diverse pocket types. Furthermore, the presence of allosteric effects, where binding events in one pocket can influence the properties of another, challenges common AI architectures that typically process local regions independently. This long-range dependency requires sophisticated neural network architectures capable of capturing correlations between spatially distant regions of the protein structure.
Table 2 categorizes key binding pocket features by their static and dynamic representations, highlighting how these characteristics must be encoded for AI modeling. The classification spans from basic volumetric measures to complex physicochemical properties, emphasizing the computational challenges in capturing each feature’s temporal evolution.
Table 2: Key structural features into their static and dynamic components
| Feature Type | Static Representation | Dynamic/Temporal Representation | Challenges in Representation |
| Pocket volume | Single scalar value (Single value, e.g., [500] ų) |
Time-varying volume measurements (Time series matrix like [500, 520, 480, 510] ų) |
Definition of pocket boundaries changes |
| Surface residues | Binary pocket/non-pocket classification (Binary vector like [0,1,0,1]) |
Dynamic exposure scores (Probability matrix like [0.2, 0.8, 0.5, 0.9]) |
Transient pocket formation |
| Physicochemical properties | Static feature vector: – H-bond donors/acceptors – Hydrophobicity scores – Electrostatic potential (Feature vector like [-1, 0.8, 2.3]) |
Time-dependent feature matrices: – Changing H-bond networks – Dynamic solvent accessibility – Fluctuating charge distributions (e.g., Time-dependent matrix [[-1.0 0.8 2.3] [-0.8 0.7 2.1] [-1.2 0.9 2.4]]) |
– Environmental dependencies – Water-mediated effects |
Post-translational modifications (PTMs) introduce an additional layer of complexity to protein representation by dynamically altering both local and global protein properties. The computational challenge lies not just in representing the chemical modifications themselves, but in capturing their effects on protein dynamics and binding site characteristics. PTMs can drastically alter the electrostatic landscape, introduce new hydrogen bond networks, and modify hydrophobic surfaces – all of which must be accurately reflected in scoring functions. Moreover, these modifications often operate in combinatorial patterns, where multiple PTMs can interact synergistically or antagonistically, creating an exponential explosion in the number of possible protein states that must be considered in virtual screening workflows.
The distinction between allosteric and orthosteric sites presents unique challenges for computational representation and scoring. While orthosteric sites typically have well-defined geometric and physicochemical properties optimized for their natural ligands, allosteric sites often exhibit more subtle and complex characteristics. The key computational challenge lies in capturing the long-range conformational coupling between allosteric and orthosteric sites, where binding events at one site can propagate through the protein structure to influence properties at distant locations. This coupling necessitates scoring functions that can account for non-local effects and protein-wide conformational changes, moving beyond traditional additive scoring terms to capture the cooperative nature of allosteric regulation. Furthermore, allosteric sites often display greater conformational plasticity than orthosteric sites, requiring more sophisticated representation schemes that can capture their dynamic nature.
While traditional virtual screening focuses primarily on well-defined binding pockets, protein-protein interaction (PPI) interfaces present distinct computational challenges that push the boundaries of current scoring approaches. These interfaces typically involve larger surface areas with shallower geometric features and more diffuse interaction patterns compared to conventional binding sites. The computational representation must capture subtle surface complementarity, distributed hydrophobic patches, and networks of water-mediated interactions that characterize these interfaces. Moreover, these regions often lack the clear spatial boundaries of traditional pockets, making it difficult to define discrete regions for scoring calculations.
The integration of these diverse binding scenarios – from traditional pockets to PPI interfaces – into unified scoring frameworks remains a significant challenge in computational drug discovery. Current scoring functions often struggle to balance the different physical requirements and interaction patterns characteristic of each binding mode. This challenge is compounded by the need to account for varying levels of flexibility, different scales of conformational changes, and the potential for induced-fit effects across these different binding paradigms. Furthermore, the emergence of new therapeutic modalities targeting these non-traditional sites requires scoring approaches that can adapt to novel binding mechanisms while maintaining accuracy for conventional drug-like compounds.
The challenges outlined above continue to test even the most sophisticated AI models, primarily due to the fundamental tension between the complex, dynamic nature of protein systems and our current computational representations. Modern deep learning architectures, despite their impressive capabilities in pattern recognition, often struggle with the multi-scale, context-dependent nature of protein dynamics and binding interactions. While some models excel at capturing local geometric features or specific types of molecular interactions, they frequently falter when confronted with long-range conformational changes, allosteric effects, or the complex interplay between different protein regions. This disparity in performance often stems from the inherent limitations in how we represent protein structures and dynamics in machine-readable formats, where crucial information about protein flexibility, water networks, or transient states may be lost or oversimplified.
The success of AI models in protein-ligand scoring is further complicated by biases and limitations in available training data. Structural databases are often overrepresented with certain protein families and binding site types, while underrepresenting challenging cases like PPI interfaces or allosteric sites. Additionally, the static nature of most crystallographic data fails to capture the dynamic aspects of protein-ligand interactions, leading to potential blind spots in model training. These challenges are exacerbated by the difficulty in obtaining negative examples and the inherent noise in experimental binding data. Moreover, the choice of molecular representation and featurization methods can significantly impact model performance, with different approaches showing varying degrees of success depending on the specific protein system and binding scenario being studied. This suggests that future advances may require not just more sophisticated AI architectures, but fundamentally new ways of representing and encoding protein structural information.
Neuralgap Genesys is a multimodal transformer based model that uses our proprietary multi-unimodal architecture to leverage the dynamic receptor-ligand data and predict bioactivity.
©2023. Neuralgap.io