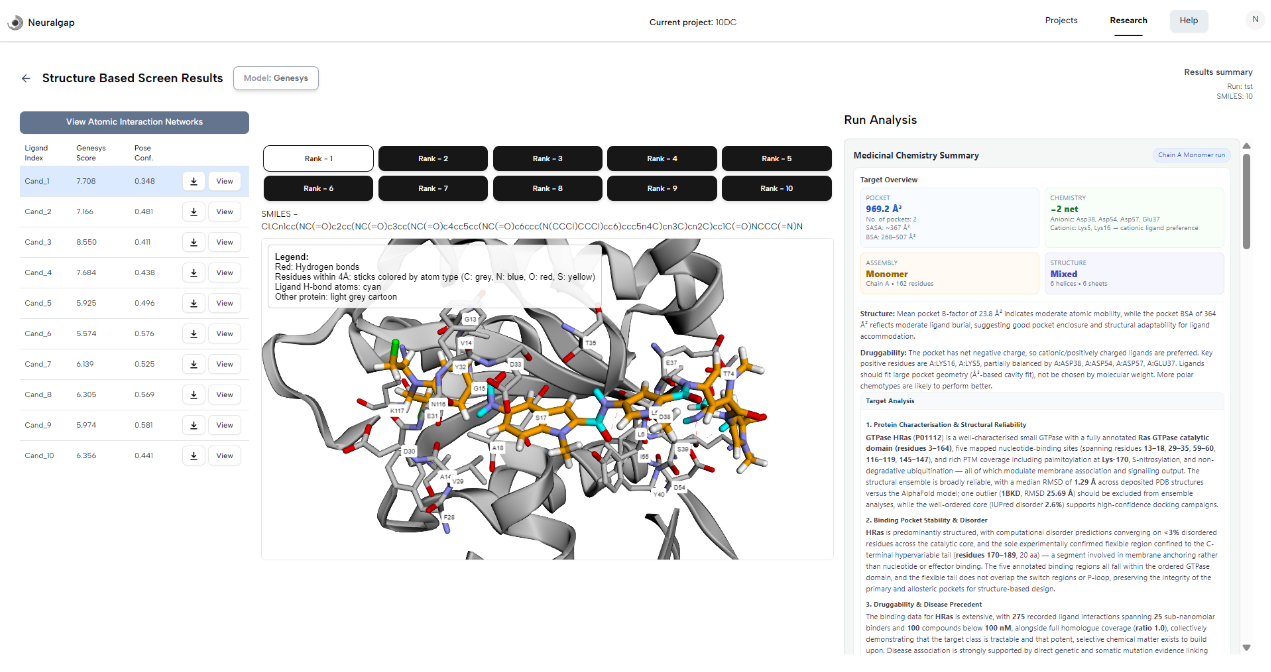
Architecture
- Unified encoding of protein, peptide, and small-molecule chemotypes including close analogs
- Multi-Unimodal (MUM) training for incremental extension across modalities without full retraining
- Latent organisation over protein / domain classes