neuralgap.io
Conventional Molecular Dynamics (cMD), which predominantly considers all atoms, is a powerful computational technique for simulating the motion and interactions of individual atoms within molecular systems. Developed in the 1970s and refined over decades, cMD offers unprecedented atomic-level detail, allowing researchers to study the dynamic behavior of complex biological molecules, materials, and chemical reactions with remarkable precision. By incorporating detailed force fields that describe interatomic interactions and solving Newton’s equations of motion for each atom, cMD can simulate larger systems over extended time scales. This capability provides invaluable insights into processes ranging from protein folding and drug-target interactions to materials properties and chemical reactions, making cMD an essential tool in fields such as biochemistry, materials science, and pharmaceutical research.
In cMD, each individual atom in a molecular system is explicitly represented and simulated. The atomic representation in cMD involves two key aspects; the level of detail in atomic modeling and the force field parameters used to describe atomic interactions. These choices directly impact the accuracy of the simulation and its computational demands.
Atomic Detail and Computational Demands
In cMD, the level of detail is fixed at the atomic scale, with each atom treated as a distinct particle. This granularity allows for the most accurate representation of molecular systems, capturing subtle effects such as hydrogen bonding, π-π stacking, and local electronic effects. However, this high level of detail comes at a significant computational cost. cMD simulation has to capture the spatial and temporal scales of simulations. On a spatial scale cMD needs to accurately model systems up to hundreds of thousands of atoms, (but struggles with larger systems like entire cellular organelles or material bulk properties), and on a temporal scale, it needs to capture fast atomic vibrations using time steps in the femtosecond range, limiting simulation times to nanoseconds or microseconds for most systems.
Element Types and Force Fields
In cMD, each atom is assigned a specific element type based on its chemical identity and local environment. These element types are crucial for defining atomic interactions. Force fields, which are sets of potential energy functions and parameters, describe the interaction parameters for different element types. They model various types of interactions, including bonded interactions (such as bond stretching, angle bending, and dihedral rotations) and non-bonded interactions (like van der Waals forces and electrostatic interactions).
Biomolecular force fields, such as AMBER, CHARMM, and OPLS-AA, are optimized for proteins, nucleic acids, and other biological molecules. For a wider range of organic molecules, general-purpose force fields like GAFF and CGenFF are employed. Additionally, specialized force fields exist for inorganic systems, materials, and specific classes of compounds.
The choice of force field is critical in cMD and depends on the system being studied. Selecting an appropriate force field directly impacts the accuracy and reliability of simulation results. Researchers must carefully consider their system of interest and the specific properties they wish to study when choosing a force field for their cMD simulations.
Parameterization involves assigning specific parameters to each atom and interaction in the system, ensuring that the simulation accurately represents the physical and chemical properties of the molecules being studied. This process is more complex and detailed in cMD compared to coarse-grained models due to the atomic-level resolution.
Force Field Selection and Customization
The foundation of parameterization in cMD is the choice of force field. Building on our introduction to force fields, the process of selecting and customizing them involves several key steps. Researchers must first evaluate available force fields based on their system of interest, reviewing literature, benchmarks, and validation studies to determine which best reproduces experimental data for similar systems. Often, a single force field may not cover all molecule types in a complex system. For instance, a protein-ligand system might require a combination of a biomolecular force field for the protein and a general-purpose force field for the ligand, necessitating careful consideration of force field compatibility. In cases where existing force fields inadequately describe certain molecular interactions, researchers may need to refine or develop new parameters. This refinement process can involve quantum mechanical calculations, fitting to experimental data, or even machine learning approaches.
Atomic Charge Assignment
A crucial aspect of parameterization in cMD is the assignment of partial atomic charges, which govern electrostatic interactions:
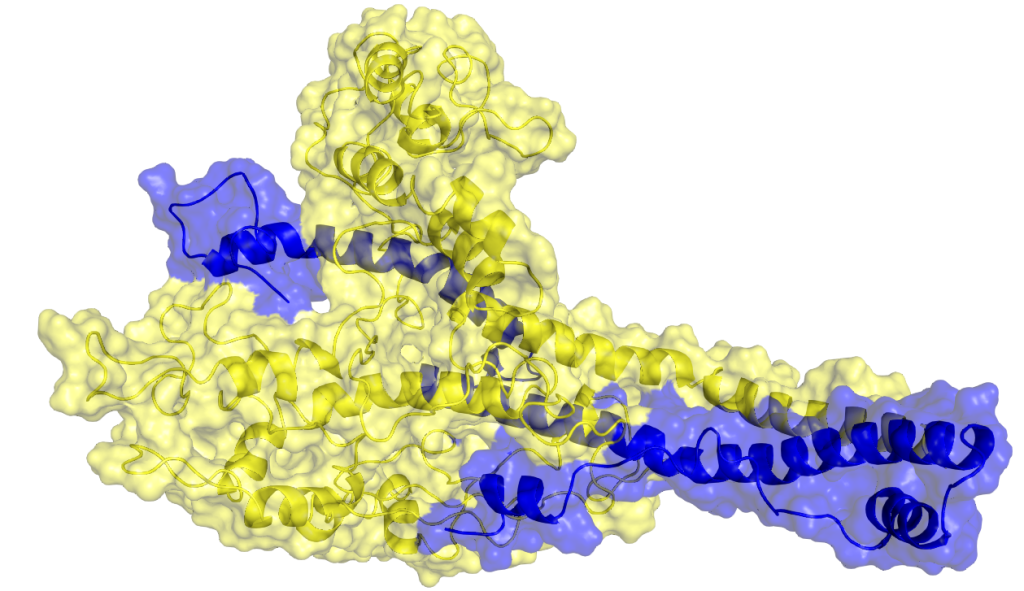
A protein heterodomer interaction (cMyc/Max) that was studied by utilising MD simulations. c-Myc and Max proteins are shown in yellow and blue, respectively
Of course, the above discussion isn’t exhaustive. cMD parameterization often involves addressing special cases such as metal ions and post-translational modifications in biological systems. Metal ions, especially transition metals, present significant challenges due to their complex electronic structures and coordination geometries. Similarly, novel compounds, including newly synthesized molecules or those not well-represented in existing force fields, necessitate comprehensive parameterization. These cases typically require a combination of quantum mechanical calculations, experimental data fitting, and validation simulations to ensure accurate representation in cMD simulations.
At the heart of cMD simulations are the interatomic forces that dictate the motion and behavior of individual atoms. Understanding these forces is crucial for interpreting simulation results and recognizing the strengths and limitations of cMD models. In contrast to coarse-grained models, cMD captures a wider range of interactions with greater detail, providing a more complete picture of molecular behavior at the atomic scale.
Bonded Interactions
Bonded interactions model forces between atoms directly connected by chemical bonds, maintaining molecular structure and flexibility in cMD simulations. Four main types are:
Non-bonded Interactions
Non-bonded interactions govern the forces between atoms not directly connected by chemical bonds. These interactions are crucial for accurately modeling molecular behavior and structure in cMD simulations.
While modeling interatomic forces forms the foundation of cMD, the real power of these simulations lies in translating microscopic interactions into observable macroscopic properties and dynamic behaviors. Here are key applications and outcomes of cMD simulations:
In conclusion, cMD simulations offer unprecedented insights into molecular behavior by precisely modeling atomic interactions and translating them into observable properties. As computational power and methodologies continue to advance, cMD will play an increasingly crucial role in fields ranging from drug discovery to materials science.
Stay tuned for our next article, where we’ll explore its sister technique, coarse-grained molecular dynamics, and how it complements cMD in tackling larger-scale biological and materials systems!
Neuralgap helps Biotech Startups bring experimental AI into reality. If you have an idea - but need a team to rapidly iterate or to build out the algorithm - we are here.
©2023. Neuralgap.io